What happens around an Alzheimer plaque?
The brains of people living with Alzheimer’s are riddled with plaques: protein aggregates consisting mainly of amyloid beta. Despite decades of research, the real contribution of these plaques to the disease process is still not clear. A research team led by Bart De Strooper and Mark Fiers at the VIB-KU Leuven Center for Brain & Disease Research in Leuven, Belgium used pioneering technologies to study in detail what happens in brain cells in the direct vicinity of plaques. Their findings, published in the prestigious journal Cell, show how different cell types in the brain work together to mount a complex response to amyloid plaques which is likely protective at first, but later on damaging to the brain.
The role of amyloid plaques in Alzheimer’s disease has puzzled scientists ever since Alois Alzheimer first described them in the brain of a woman with young onset dementia. Now, over a century later, we have learned a lot about the molecular processes that lead to neurodegeneration and subsequent memory loss, but the relationship between the plaques and the disease process in the brain is still ambiguous.
“Amyloid plaques might act as a trigger or as a driver of disease, and the accumulation of amyloid beta in the brain likely initiates a complex multicellular neurodegenerative process,” says professor Bart De Strooper (VIB-KU Leuven).
His team set out to map the molecular changes that take place in cells near amyloid plaques.
“We used the latest technologies to analyze genome-wide transcriptomic changes induced by amyloid plaques in hundreds of small tissue domains,” explains Mark Fiers, co-lead on the study. “In this way, we could generate a large data set of transcriptional changes that occur in response to increasing amyloid pathology, both in mouse and human brains.”
Two co-expression networks
Picture: In the brains of transgenic mice aged 3, 6, 12 and 18 months, amyloid plaques (white) elicit a response from astrocytes, a specific type of brain cells, colored in green.
“We focused on the transcriptomic changes in the immediate neighborhood of the amyloid plaques, with a 50 micrometer perimeter,” explains Wei-Ting Chen, a postdoc in De Strooper’s team.
In a well-studied genetic mouse model showing amyloid pathology, the scientists identified two novel gene co-expression networks that appeared highly sensitive to amyloid beta deposition.
Chen: “With increasing amyloid beta deposition, a multicellular co-expressed gene response was established encompassing no less than 57 plaque-induced genes.” These genes were mainly expressed in astroglia and microglia, two types of supportive brain cells, and were not co-expressed in the absence of amyloid plaques.
“We also found interesting alterations in a second network, expressed mainly by another type of cells, namely oligodendrocytes,” adds Ashley Lu, PhD student in the team. “This gene network was activated under mild amyloid stress but depleted in microenvironments with high amyloid accumulation.”
“Many of the genes in both networks show similar alterations in human brain samples, strengthening our observations,” adds Fiers.
Targeting plaques
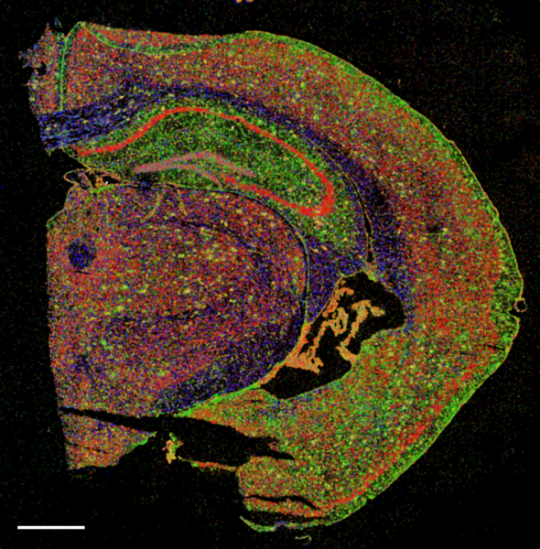
Picture: A section of half a mouse brain in which different types of brain cells are stained.
“Our data demonstrate that amyloid plaques are not innocent bystanders of the disease, as has been sometimes suggested, but in fact induce a strong and coordinated response of all surrounding cell types,” says De Strooper. “Further work is needed to understand whether, and when, removal of amyloid plaques—for instance by antibody therapy currently in development to treat amyloid plaques—is sufficient to reverse these ongoing cellular processes.”
Whether antibody binding to amyloid plaques could also modulate these glial responses remains to be determined.
“It would in any case complicate the interpretation of the outcome of clinical trials as these cellular effects might be different between different antibodies,” adds De Strooper.
Publication
Spatial transcriptomics and in situ sequencing uncover coordinated cell responses to amyloid-β plaques in Alzheimer’s disease, Chen, Lu et al. Cell 2020
DOI: 10.1016/j.cell.2020.06.038